1) Mukopolisakkaridoz hastalığı nedir?
Mukopolisakkaridoz (MPS) hastalıkları, doğuştan gelen, birçok sistemi etkileyen, genetik geçişli metabolizma hastalığıdır. Glikozaminoglikanların (GAG) yıkımında görevli enzimlerin eksiklikleri sonucu oluşur ve GAG birikimi ile karakterize lizozomal depo hastalığıdır.
2) Ne sıklıkta görülür?
Avrupa’da yapılan çalışmalarda MPS hastalığının sıklığı 1.56-4.8:/100000 olarak bildirilmektedir. Ülkemizde hastalığın insidansı tam olarak bilinmemektedir.
3) Genetik geçişi nasıl olur?
MPS tip II hariç diğer tiplerin genetik geçişi otozomal çekinik dediğimiz kalıtım tipine uyar. Bu kalıtım tipinde, anne ve babanın her ikisi de hastalığın taşıyıcısıdır. Her ikisi de taşıyıcı olan anne-babanın çocukları, taşıyıcı, hasta ya da sağlıklı olabilir. Bir ailede hasta bir çocuk doğduysa, hastalığın her kardeşte tekrarlama riski vardır. Bu nedenle hasta bir çocuğu olan ailelere daha sonraki çocuklar için mutlaka doğum öncesi genetik tanı yaptırmaları önerilmektedir. MPS II ise X’ e bağlı resesif kalıtım gösterir. Nadir durumlar dışında hastalık erkeklerde saptanır. Tanı alan hastaların aileleri mutlaka genetik danışmanlık verilir.
4) Hastalık vücutta ne gibi etkiler yapar?
MPS hastalarında dokularda GAG birikimine bağlı değişiklikler sonucu klinik bulgular oluşmaktadır. GAG hücre dışı matriksin önemli bir bileşenidir. Birçok biyolojik olayda görev almaktadır. GAG anormal birikimi dokuların normal yapısının bozulmasına ve fonksiyon kaybına yol açmaktadır. MPS hastalarında birçok organı etkileyen tutulum söz konusudur.
5) Klinik bulguları nelerdir?
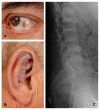
Mukopolisakkaridozlar kronik, ilerleyici ve çoklu sistem tutulumu ile karakterize lizozomal depo hastalıklarıdır. Genelde hastalar doğumda normaldir. Zamanla klinik bulgular ortaya çıkmaktadır. Klinik bulgular hafif ya da ağır formda karşımıza çıkabilir. MPS tipine göre klinik bulgular değişkenlik göstermektedir. MPS genel olarak görülen klinik bulgular arasında kaba yüz görünümü, zihinsel yetersizlik, gelişme geriliği, işitme kaybı, gözde korneal bulutlanma, karaciğer, dalak büyüklükleri, sık tekrarlayan solunum yolu enfeksiyonları, adenoid hipertrofi, dil büyüklüğü, inguinal ve umblikal herniler, akciğer hastalıkları, kalp hastalıkları (kas, kapak tutulum ritim problemleri) eklem katılıkları, boy kısalığı, kemik bulguları (dizostosis mültipleks), uyku apne sendromu yer almaktadır.
6) Hangi tipleri vardır?
Kendi içinde 7 ana alt gruba ayrılır; MPS I, II, III, IV, VI, VII, IX, X. Kendi içinde alt gruplara ayrılmaktadır. MPS I (Hurler, Hurler-Scheie- Scheie) , MPS III (A,B,C ve D)
MPS I (Hurler, Hurler-Scheie, Scheie) Alfa L-iduronidaz eksikliği sonucu oluşmaktadır. Heparan sülfat ve dermatan sülfat birikimi olur. Hurler formu ağır formudur ve genellikle ilk 1 yaşta tanı alır. Kaba yüz görünüm, boy kısalığı, hepatosplenomegali, korneal bulutlanma, ilerleyici öğrenme güçlüğü, kalp hastalığı, kemik bulguları sık görülen klinik bulgulardır. Scheie form hafif klinikl gösterir, entelektüel olarak normaldirler, normale yakın boyları vardır.
MPS II (Hunter hastalığı)
İduronat 2 sulfataz eksikliği sonucu oluşur. Klinik bulgular MPS I Hurler benzeridir.Korneal etkilenme görülmeyebilir. Nörolojik tutulum belirgin olmadığı nonnöronopatik formu ve ara formları vardır.
MPS III (Sanflippo Hastalığı)
Kendi içinde 4 alt gruba ayrılır. MPS IIIA heparan N sulfataz, MPS IIIB N asetil-glukozaminidaz, MPS IIIC Asetil CoA glukozamin N-asetiltransferaz, MPS IIID N asetil glukozamin 6 sulfataz ensim eksikliği sonucu oluşur. Santral sinir sistem tutulumu belirgindir.
MPS IVA (Morquio Hastalığı)
Galaktoz-6-sulfataz eksikliği sonucu oluşmaktadır. Ağır iskelet sistemi tutulumu vardır. Entelektüel bozukluk gözlenmez. Daha hafif klinik seyirli atenüe formu da mevcuttur. Eklemlerde hiperlaksisite mevcuttur.
MPS VI ( Maroteaux-Lamy Hastalığı)
N-asetil galaktozamin-4- sulfataz enzim eksiliği sonucu oluşur. Nörolojik bozukluk gözlenmez. İskelet tutulum, boy kısaslığı, eklem tutulum sık görülür. Kalp, hastalığığı, işitme kaybı, göz tutulum, herni, uyku apne sendromu sık görülür.
MPS VII ( Sly Hastalığı)
β-glukuronidaz enzim eksikliği sonucu oluşur. Non-immun hidrops fetalis sık rastlanan klinik bulgudur. Atenüe form MPS I,II ve VI klinik bulguları ile benzerlik gösterir.
MPS IX
Hyaluronidaz enzim eksikliği sonucu oluşur. Çok nadir görülür. Boy kısalığı ve Juvenil idiopatik artrit benzeri klinik bulgular görülür
MPS X
Arilsulfataz K (ARSK) eksikliği sonucu oluşur
7) Tanısı nasıl konur?
Metabolizma uzmanı, hastanın öyküsü, muayenesi ve laboratuvar tetkikleri ile Mukopolisakkaridoz hastalığı için ön tanıyı koyar. İdrarda GAG analizi, enzim düzeylerinin tayini tanı için kullanılır. Kesin tanı için genetik analiz yapılır.
8) Hangi tetkikler ne sıklıkta yapılmalıdır?
Hastalar düzenli aralıklarla klinik olarak değerlendirilir, fizik muayene ve hastalığın tutulan organ sistemleri açısından gerekli tetkikler ile takip edilir. Kan, idrar testleri, karaciğer, dalak görüntülenmesi, göz, işitme muayeneleri, kemik değerlendirilmeleri, kalp ve akciğer tutulum açısından taramaları, beyin ve omurilik görüntüleme incelemeleri, gelişim testleri ile incelenir. Çoklu organ tutulumu nedeni ile farklı branş hekimleri ile ortak izlem yapılmaktadır. Takip sıklığı hastalığın şiddetine göre değişkenlik gösterebilir. Genel olarak organ tutulumları açısından 6 ay- 1 yıl aralarla değerlendirilmeleri gerekir. Hastanın durumuna göre takip sıklığı daha kısa aralıklarla yapılabilmektedir.
9) Tedavi seçenekleri nelerdir?
MPS hastalarında tedaviye erken başlanması önemlidir. MPS hastalığın tiplerine göre farklı tedavi seçenekleri söz konusudur. Günümüzde eksik olan enzimin yerine konması olan Enzim replasman tedavisi MPS, I, II, IV, VI ve VII hastaları için yapılabilmektedir. MPS I için kök hücre nakli seçili hastalarda uygulanabilen tedavi yöntemidir. MPS hastalarında tutulan organlara yönelik tıbbi ve cerrahi girişimler tedavi olarak uygulanmaktadır. Nörolojik tutulum gösteren hastalarda santral sinir sistemine etkili ilaç çalışmaları devam etmektedir.
10) Uzun dönemde hangi sorunlar ile karşılaşabiliriz? Mukopolisakkaridozlarda hastalık tipine göre, uzun dönemde, nörolojik tutulumun ilerlemesine bağlı sorunlar (yeti kaybı, nöbetler, beslenme, uyku problemleri gibi), cerrahi gerektirecek iskelet problemleri, ilerleyici görme ve işitme problemleri, ilerleyici kalp ve hastalıkları görülebilmektedir.